Archive: What’s in a Number? Estimating Microbial Richness using DADA2
February 17th, 2020
Corinne Walsh & Noah Fierer
Soils are often touted as harboring some of the most diverse microbial communities of any habitat on Earth. We typically expect to detect on the order of thousands of different bacterial taxa in a given soil sample using culture-independent sequencing methods. However, richness (number of distinct taxa detected per sample) is arguably the community metric that is most susceptible to methodological differences such as sequencing depth (and platform), rarefaction or thresholding cut-offs, and even choice of bioinformatic pipeline. We recently encountered an intriguing discrepancy in richness estimates due to a single bioinformatic parameter, and thought our ponderings would be worthwhile to share with others that have encountered, or may encounter, similar discrepancies.
To illustrate how different data processing methods can yield drastically different richness estimates, we use an example encountered when running the DADA2 pipeline (paper) to process bacterial and archaeal 16S rRNA gene sequence data (our lab’s workflow here). Using the standard pipeline where individual samples are processed independently and in parallel (samples 'unpooled'), we were estimating richness levels of around several hundred ASVs (amplicon sequence variants) per soil sample. This was somewhat surprising as we were expecting to observe far more ASVs per soil sample at a comparable sequencing depth. This discrepancy in richness estimates has held true across various soil datasets in the lab, and has inspired a friendly debate about richness interpretations, and data expectations more generally. Why are richness estimates obtained using the standard DADA2 workflow seemingly so low and, perhaps more importantly, is this a problem?

We compared a few different pipelines and function parameters to learn what step of the bioinformatic processing was contributing to the observed difference in richness values. We found that changing a parameter in DADA2’s sequence inference function (“dada()”) to run all samples pooled together (pool = TRUE, 'pooled') resulted in sample richness numbers almost FOUR-FOLD HIGHER than when samples were processed in parallel (pool = FALSE, 'unpooled') (Fig 1). In a test dataset of 34 soil samples, the total ASV counts across all 34 samples remained similar across processing methods ('unpooled' = 17454 ASVs, 'pooled' = 17179 ASVs), but the number of ASVs per sample was notably higher using the 'pooled' method ('unpooled' = 767 ASVs; 'pooled' = 2867) (Fig 2). This difference cannot be attributed to read counts, as total read counts across the entire sample set were similar ('unpooled' = 722935 reads; 'pooled' = 825188 reads) and average per-sample read counts were similar ('unpooled = 21262; 'pooled' =24270) regardless of whether samples were processed together or independently. An obvious consequence of this richness difference is that sample beta diversity goes down (more ASVs shared between samples), and alpha diversity (# of ASVs per sample) goes way up when using the 'pooled' method compared to the 'unpooled' method. In other words, the 'pooled' and 'unpooled' options for identifying ASVs led to dramatically different richness estimates with far fewer ASVs detected per sample using the 'unpooled' option, but similar total numbers of ASVs across an entire set of samples.

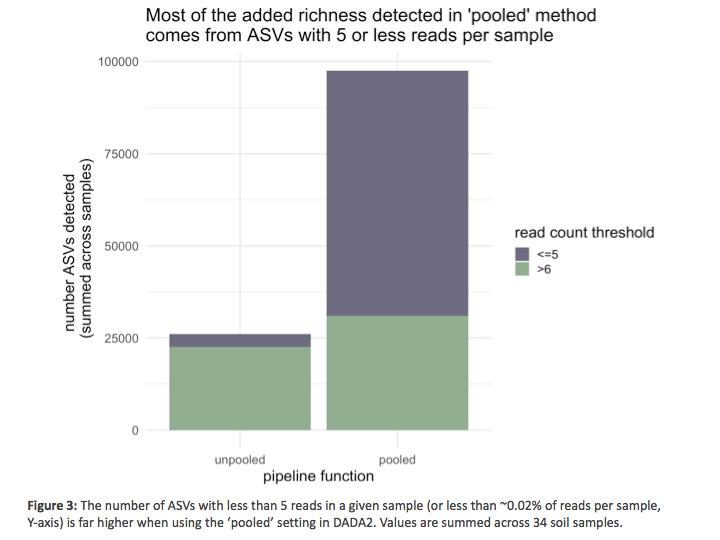
Further investigation revealed that the vast majority of these additional ASVs detected using the 'pooled' method are rare, with most of the added ASVs detected per sample having < 5 reads in a given sample (Fig 3). SO, while the pooling of samples does result in higher richness values, most of this added richness comes from relatively rare taxa (in this case individual ASVs that typically account for <0.02% of reads per sample). DADA2’s creator Ben Callahan states in his tutorial that using this pooling parameter does increase sensitivity towards rare taxa (see here for more detail, and here for even more discussion). We understand this to be due to the challenge of distinguishing between sequencing error & biological reality at really low read counts when samples are processed independently (unpooled) - a challenge that is mitigated if the sequence variants are identified when pooling the data across many samples. This may not be surprising to anyone familiar with DADA2, but it is still worth noting, ASVs that are rare in individual samples may not be identified when analyzing samples individually (the 'unpooled' setting) because the low read counts per sample make it difficult, if not impossible, for the algorithm to distinguish between sequencing error and real, biological variation in the amplicon sequences.
SO WHAT ARE WE GOING TO DO?!
Ultimately the choice of function parameter ('pooled' vs. 'unpooled') depends on your sample type and scientific questions. If you have a relatively simple community (10 to low 100s of taxa) or don’t care about rare taxa, running samples 'unpooled' is a good choice because the richness discrepancy is negligible in less diverse communities, and computational time is SIGNIFICANTLY shorter with the 'unpooled' method. However, in complex communities where we see large differences in richness, pooling samples does allow for more sensitive detection of rare taxa that may be commonly observed across multiple samples. While compute time is substantially higher, running samples pooled together results in more ASVs detected per sample, from which the user can make tailored decisions for how to estimate richness. We should note that there are some great resources out there for calculating and simultaneously characterizing the UNCERTAINTY of microbial richness estimates, such as the breakaway R package from Amy Willis of the statistical diversity lab (also see here for a paper on methods for estimating richness in complex communities).
SO WHAT DOES THIS ALL MEAN?!
As previously stated, there are many reasons to not trust richness data (among them sampling depth, processing methods, etc). While in many cases it can be useful to have a rough estimate for the number of taxa found in a given sample, richness is not necessarily a parameter that is that is particularly interesting or critical to many ecological questions and studies (a good perspective on this here). Additionally, we know that well-to-well contamination likely occurs more than any of us would care to admit, and such contamination could be contributing to some of the rare taxa consistently detected at low read counts across multiple samples (see paper, blogpost for more). We think it is important to consider what you actually want to know when estimating richness, calculate the uncertainty around those estimates, and keep in mind the significant caveats when interpreting richness estimates (e.g. consider read depth and thresholds of detection, don’t compare across studies). As a general rule of thumb for amplicon sequence datasets, beware of super rare taxa and don’t put too much weight in stand-alone richness metrics.